
Mechanism of glycoside hydrolysis: A comparative QM/MM molecular dynamics analysis for wild type and Y69F mutant retaining xylanases†
Abstract
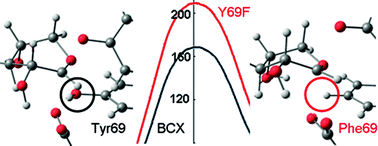
Computational simulations have been performed using hybrid quantum-mechanical/molecular-mechanical potentials to investigate the catalytic mechanism of the retaining endo-β-1, 4-xylanase (BCX) from B. circulans. Two-dimensional potential-of-mean-force calculations based upon molecular dynamics with the AM1/OPLS method for wild-type BCX with a p-nitrophenyl xylobioside substrate in water clearly indicates a stepwise mechanism for glycosylation: the rate-determining step is nucleophilic substitution by Glu78 to form the covalently bonded enzyme-substrate intermediate without protonation of the leaving group by Glu172. The geometrical configuration of the transition state for the enzymic reaction is essentially the same as found for a gas-phase model involving only the substrate and a propionate/propionic acid pair to represent the catalytic glutamate/glutamic acid groups. In addition to stabilizing the 2,5B boat conformation of the proximal xylose in the non-covalent reactant complex of the substrate with BCX, Tyr69 lowers the free-energy barrier for glycosylation by 42 kJ mol−1 relative to that calculated for the Y69F mutant, which lacks the oxygen atom OY. B3LYP/6-31+G* energy corrections reduce the absolute height of the barrier to reaction. In the oxacarbenium ion-like transition state OY approaches closer to the endocyclic oxygen Oring of the sugar ring but donates its hydrogen bond not to Oring but rather to the nucleophilic oxygen of Glu78. Comparison of the average atomic charge distributions for the wild-type and mutant indicates that charge separation along the bond between the anomeric carbon and Oring is matched in the former by a complementary separation of charge along the OY–HY bond, corresponding to a pair of roughly antiparallel bond dipoles, which is not present in the latter.
 Please wait while we load your content...
Please wait while we load your content...

