
A total synthesis of milbemycin G: approaches to the C(1)–C(10)-fragment and completion of the synthesis
Abstract
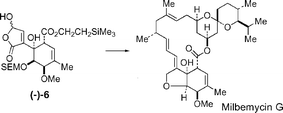
A synthesis of the hydroxybutenolide (−)-6 required for synthesis of α-milbemycins and the completion of a total synthesis of milbemycin G 7 is reported.
Following preliminary studies, an optimised synthesis of the hydroxybutenolide (−)-6 from the hydroxyketone 38 was developed which involved the resolution of 38 by separation of the 3-(O-chloroacetyl)-(S)-mandelates 80 and 83. Ester 80, which corresponded to the required enantiomer of the hydroxyketone 38, crystallized from the mixture of the diastereoisomeric esters 80 and 83 giving the (−)-hydroxyketone (−)-38 in an overall yield of 47% (based on racemic 38) after ethanolysis. Hydroxyketone (−)-38 was oxidised to the enolic diketone (−)-39 and phenylselenation and stereoselective reduction gave the trihydroxycyclohexyl selenide (−)-43. The regioselective introduction of the non-conjugated double-bond into the six-membered ring was then achieved by esterification of the 4-hydroxyl group using trichloroacetic acid to give the trichloroacetate (−)-69. Oxidative elimination from the trichloroacetate using tert-butyl hydroperoxide was highly regioselective and gave the endo- and exocyclic alkenes (−)-44 and (−)-46 in a ratio of 95 : 5 after ethanolysis of the trichloroacetates. Selective O-methylation of the 4-hydroxyl group via the cyclic stannylene 55 and protection of the 3-hydroxyl group as its 2-trimethylsilylethoxymethyl (SEM) ether gave the ester (−)-57. Following saponification of the ethyl ester, re-esterification using 2-trimethylsilylethanol and oxidation of the 2-trimethylsilylfuryl fragment using singlet oxygen gave the required hydroxybutenolide (−)-6.
The Wittig reaction between the phosphonium salt 2 and the hydroxybutenolide (−)-6 gave a ca. 2 : 1 mixture of the (4Z)- and (4E)-isomers of the ester 84 which on treatment with a catalytic amount of iodine was converted into the (4E)-isomer (4E)-84. Deprotection gave the seco-acid 85 but attempts to macrocyclise this were unsuccessful, the elimination product 86 being the only product isolated. The Wittig product 84 was taken through to the butenolide (2′E)-91 by removal of the SEM group, cyclisation to form the butenolide ring and diene isomerization, but this could not be converted into the corresponding seco-acid 92. However, removal of the SEM group from the seco-acid 85 gave the trihydroxy-acid 93 which was cyclized under modified Yamaguchi conditions to give the macrolide 94 together with a small amount of the macrocyclic butenolide 95. Reduction of this mixture using diisobutylaluminium hydride gave (6R)-6-hydroxymilbemycin E 96 which was converted to milbemycin G 7 by cyclisation of the primary chloride 97. The synthetic milbemycin G 7 was identical to a sample prepared by methylation of a commercial sample of milbemycin D 98, 7-O-methylmilbemycin G 99 being a side-product of this methylation.
 Please wait while we load your content...
Please wait while we load your content...

