
Tuning the electronic structure of transition metals embedded in nitrogen-doped graphene for electrocatalytic nitrogen reduction: a first-principles study†

Abstract
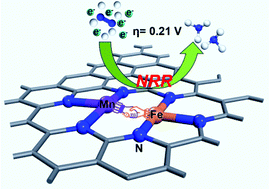
As one of the most important subjects in chemistry, nitrogen activation and reduction to yield ammonia is still a big challenge. The lack of deep understanding of the nitrogen reduction reaction (NRR) impedes the development of high-performance catalysts. In the present study, we introduce a second transition metal (M = Mn, Fe, Co, Ni, Cu, Zn, and Mo) into the active site of a single-atom Fe–N–C catalyst to tune the electronic structure and study the activity of the as-designed neighboring bimetal Fe/M–N–C catalyst in the electrochemical NRR under acidic conditions, by performing first-principles calculations. By checking the stability of the catalysts, the adsorption ability for N2, the Gibbs free energy change for the potential-determining step in the NRR, and the hydrogen evolution reaction (HER) activity, only the Fe/Mn–N–C catalyst is predicted to be a promising candidate for the NRR as it shows significantly improved catalytic activity and strong selectivity against the HER. A mechanistic study reveals the synergistic effects of the bimetal active sites, and the introduced Mn atom generates a strong multi-reference effect on the electronic configuration to create more tunnels to transfer the d-orbital electrons to activate the inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond, inducing the “acceptance–donation” process to facilitate the activation and reduction of N2. The current results provide an effective strategy to design stable, active, and selective catalysts for the electrochemical NRR.
N triple bond, inducing the “acceptance–donation” process to facilitate the activation and reduction of N2. The current results provide an effective strategy to design stable, active, and selective catalysts for the electrochemical NRR.
 Please wait while we load your content...
Please wait while we load your content...

