
Defect-enriched tunability of electronic and charge-carrier transport characteristics of 2D borocarbonitride (BCN) monolayers from ab initio calculations†

Abstract
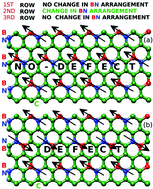
Development of inexpensive and efficient photo- and electro-catalysts is vital for clean energy applications. Electronic and structural properties can be tuned by the introduction of defects to achieve the desirable electrocatalytic activity. Using first-principles molecular dynamics simulations, the structural, dynamical, and electronic properties of 2D borocarbonitride (h-BCN) sheets have been investigated, highlighting how anti-site defects in B and N doped graphene significantly influence the bandgap, and thereby open up new avenues to tune the chemical behavior of the 2D sheets. In the present work, all of the monolayers investigated display direct bandgaps, which reduce from 0.99 eV to 0.24 eV with increasing number of anti-site defects. The present results for the electronic structure and findings for bandgap engineering open up applications of BCN monolayers in optoelectronic devices and solar cells. The influence of the anti-site distribution of B and N atoms on the ultra-high hole/electron mobility and conductivity is discussed based on density functional theory coupled with the Boltzmann transport equation. The BCN defect monolayer is predicted to have carrier mobilities three times higher than that of the pristine sheet. The present results demonstrate that BN doped graphene monolayers are likely to be useful in the next-generation 2D field-effect transistors.
- This article is part of the themed collections: Editor’s Choice: Computational studies of nanomaterials for energy, catalysis and electronics, Advisory Board research selection and Nanoscale 10th Anniversary Special Issue
 Please wait while we load your content...
Please wait while we load your content...

