
From monomer to monolayer: a global optimisation study of (ZnO)n nanoclusters on the Ag surface†
 b
and
b
and
Abstract
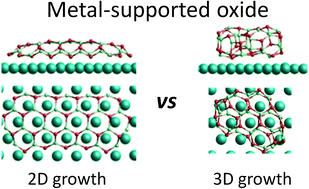
We employ global optimisation to investigate how oxide nanoclusters of increasing size can best adapt their structure to lower the system energy when interacting with a realistic extended metal support. Specifically, we focus on the (ZnO)@Ag(111) system where experiment has shown that the infinite Ag(111)-supported ZnO monolayer limit corresponds to an epitaxially 7 : 8 matched graphene-like (Zn3O3)-based hexagonal sheet. Using a two-stage search method based on classical interatomic potentials and then on more accurate density functional theory, we report global minina candidate structures for Ag-supported (ZnO)n cluster with sizes ranging from n = 1–24. Comparison with the respective global minina structure of free space (ZnO)n clusters reveals that the surface interaction plays a decisive role in determining the lowest energy Ag-supported (ZnO)n cluster structures. Whereas free space (ZnO)n clusters tend to adopt cage-like bubble structures as they grow larger, Ag-supported (ZnO)n clusters of increasing size become progressively more like planar cuts from the infinite graphene-like ZnO single monolayer. This energetic favourability for planar hexagonal Ag-supported clusters over their 3D counterparts can be partly rationalised by the ZnO–Ag(111) epitaxial matching and the increased number of close interactions with the Ag surface. Detailed analysis shows that this tendency can also be attributed to the capacity of 2D clusters to distort to improve their interaction with the Ag surface relative to more rigid 3D bubble cluster isomers. For the larger sized clusters we find that the adsorption energies and most stable structural types appear to be rather converged confirming that our study makes a bridge between the Ag-supported ZnO monomer and the infinite Ag-supported ZnO monolayer.
 Please wait while we load your content...
Please wait while we load your content...

