
The search for the most stable structures of silicon–carbon monolayer compounds†
Abstract
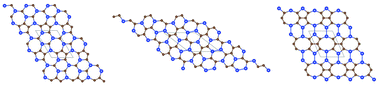
The most stable structures of two-dimensional (2D) silicon–carbon monolayer compounds with different stoichiometric compositions (i.e., Si : C ratio = 2 : 3, 1 : 3 and 1 : 4) are predicted for the first time based on the particle-swarm optimization (PSO) technique combined with density functional theory optimization. Although the 2D Si–C monolayer compounds considered here are rich in carbon, many of the low-energy metastable and the lowest-energy silicon–carbon structures are not graphene (carbon monolayer) like. Phonon-spectrum calculations and ab initio molecular dynamics simulations were also performed to confirm the dynamical stability of the predicted most stable 2D silicon–carbon structures as well their thermal stability at elevated temperature. The computed electronic band structures show that all three predicted silicon–carbon compounds are semiconductors with direct or indirect bandgaps. Importantly, their bandgaps are predicted to be close to those of bulk silicon or bulk germanium. If confirmed in the laboratory, these 2D silicon–carbon compounds with different stoichiometric compositions may be exploited for future applications in nanoelectronic devices.
 Please wait while we load your content...
Please wait while we load your content...

