
Understanding the enhanced catalytic activity of bimetallic AuCu/TiO2 in CO2 adsorption and activation: a density functional theory study†
 *ab
and
*ab
and
Abstract
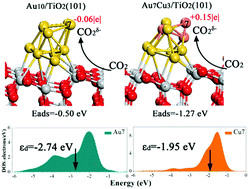
Photoreduction of CO2 to hydrocarbons has lately attracted increased attention. Understanding the reaction mechanism of photocatalytic reduction of CO2 is crucial for the development of CO2 photocatalysts. CO2 adsorption and activation are the first steps of CO2 photoreduction. In this work, the adsorption and activation of CO2 on TiO2-supported Au and Au–Cu clusters were studied using density functional theory (DFT) calculations to identify the key factors for improving its adsorption and activation. The presence of Cu atoms was found to have great significance in the adsorption and activation of CO2, and the Cu7 atom on the Au7Cu3/TiO2(101) surface exhibited the strongest CO2 adsorption ability. By analyzing the surface electronic structure, we found that the up-shift of the d-band center toward the Fermi level in the presence of Cu atoms increased the binding strength of CO2. Additionally, the Cu atoms were positively charged on the Au7Cu3/TiO2(101) surface and promoted the adsorption of CO2 by increasing the electrostatic attraction between the O atom of CO2 and the Cu atom. Therefore, compared to the Au10/TiO2(101) surface, the activity of the Au7Cu3/TiO2(101) surface can be definitely improved. Our results can provide useful insights into the design and development of more efficient bimetallic cluster supported TiO2 photocatalysts, which are used as catalysts for the synthesis of hydrocarbon fuels by CO2 hydrogenation.
 Please wait while we load your content...
Please wait while we load your content...

