
Computational characterization of N-type characteristics and optoelectronic properties in air-stable pyromellitic diimide derivatives†
Abstract
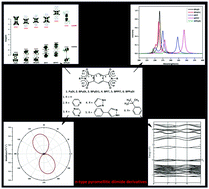
The present study investigates the charge transport and optoelectronic properties of the pyromellitic diimide derivatives BPyDI, BPyDI1, BPIT, BPPIT, and BPPyDI using density functional theory (DFT). The LUMO levels of the studied compounds (except BPPyDI and BPPIT) are found to lie in the range of −3.5 eV to −3.7 eV and the electron affinities (EAs) (except BPPyDI and BPPIT) lie in the range of 2.4 eV to 2.7 eV, suggesting that the compounds are n-type and air-stable organic semiconductors. It is noted that the principal interaction between the intermolecular layers of the crystals are mostly due to the C⋯H and H⋯H contacts and a relatively large intermolecular interaction between the molecular layers (MLs) of the contacts N⋯H, C⋯N, N⋯N and O⋯N of all the derivatives (except BPPyDI) leads to larger LUMO coupling as compared to the bare PyDI. The substitution of the functional groups is supposed to enhance the values of μeΦ and μhΦ by ∼102–103 and 101 times of the respective mobilities of the bare PyDI. Moreover, the absorption spectra of all the compounds are found to be in the UV-spectral region, particularly in the wavelength range of 244–281 nm. The bathochromic shift of the derivatives are observed in the UV region as compared to the bare PyDI compound.
 Please wait while we load your content...
Please wait while we load your content...

