
A DFT study on the mechanism and origins of the ligand-controlled regioselectivity of a palladium-catalyzed dearomatic reaction of 1-(chloromethyl)naphthalene with phenylacetonitrile†
 *b
*b
Abstract
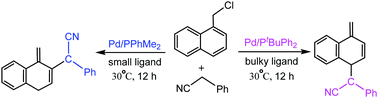
Dearomatic reactions have attracted wide attention in recent years, and they can construct important alicyclic molecules and widen the diversity of structures. Recently, Bao et al. studied the Pd-catalyzed reaction of 1-(chloromethyl)naphthalenes with (hetero)arylacetonitriles to afford ortho- or para-substituted products in high yields. The bulky ligand tBuPPh2 generates para-substituted products, while the small ligand Me2PPh provides ortho-substituted products. In this study, the density functional theory calculations were performed to investigate the mechanistic details and origins of regioselectivity. The calculations elucidated that in the case of the bulky tBuPPh2 ligand, TS4–6 (7.8 kcal mol−1) for the formation of a dearomatic para-substituted product via C–C bond coupling between the para-carbon of an η3-exo-(naphthyl)methyl ligand and the terminal carbon atom of the phenylethenimine ligand is more stable than TS4–7 (10.4 kcal mol−1) for the formation of a dearomatic ortho-substituted product, which is ascribed to the steric repulsions between the phenyl of the tBuPPh2 ligand and the phenyl of the phenylethenimine ligand. In comparison, in the case of the small Me2PPh ligand, TS4–7′ (7.3 kcal mol−1) for the formation of a dearomatic ortho-substituted product is more stable than TS4–6′ (10.2 kcal mol−1) for the formation of a dearomatic para-substituted product, which originates from the great π–π stacking interactions between the phenyl of the phenylethenimine ligand and the (naphthyl)methyl ligand.
 Please wait while we load your content...
Please wait while we load your content...

