
Metal-free homolytic hydrogen activation: a quest through density functional theory computations†
Abstract
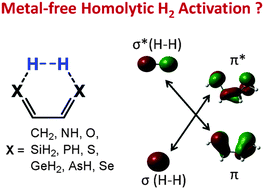
The electronic structure of s-cis-1,3-butadiene (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHCH2) encouraged us to explore a transition metal (TM) free strategy for homolytic H2 activation by replacing both terminal CH2 groups of CH2CH–CHCH2 with X = NH, O, SiH2, PH, S, GeH2, AsH, and Se. The study predicts that the six molecules with X = SiH2, PH, S, GeH2, AsH, and Se may activate H2 with barriers lower than 21.7 kcal mol−1, but the three with X = CH2, NH, and O have barriers higher than 38.9 kcal mol−1. Unlike homolytic H2 activation by TM complexes, which occurs on an active site with only one reactive center, the present activations are enabled through an active site with two reactive centers. The greatly increased reactivity of these heavier analogs originates from the significantly reduced EOFMO–EUFMO gaps (EOFMO/EUFMO represents the occupied/unoccupied frontier molecular orbital that has the correct symmetry to interact with the H2 σ*/σ orbital). Interestingly, H2 activations by experimentally reported derivatives with X = PH, S, and Se were predicted to have experimentally accessible barriers and to be exergonic, which is an exciting observation. The electronic structure of 1,3-dipoles is also suitable for designing geminal active sites for homolytic H2 activation.
CH–CHCH2) encouraged us to explore a transition metal (TM) free strategy for homolytic H2 activation by replacing both terminal CH2 groups of CH2CH–CHCH2 with X = NH, O, SiH2, PH, S, GeH2, AsH, and Se. The study predicts that the six molecules with X = SiH2, PH, S, GeH2, AsH, and Se may activate H2 with barriers lower than 21.7 kcal mol−1, but the three with X = CH2, NH, and O have barriers higher than 38.9 kcal mol−1. Unlike homolytic H2 activation by TM complexes, which occurs on an active site with only one reactive center, the present activations are enabled through an active site with two reactive centers. The greatly increased reactivity of these heavier analogs originates from the significantly reduced EOFMO–EUFMO gaps (EOFMO/EUFMO represents the occupied/unoccupied frontier molecular orbital that has the correct symmetry to interact with the H2 σ*/σ orbital). Interestingly, H2 activations by experimentally reported derivatives with X = PH, S, and Se were predicted to have experimentally accessible barriers and to be exergonic, which is an exciting observation. The electronic structure of 1,3-dipoles is also suitable for designing geminal active sites for homolytic H2 activation.
 Please wait while we load your content...
Please wait while we load your content...

