
Combining electronic and structural features in machine learning models to predict organic solar cells properties†

Abstract
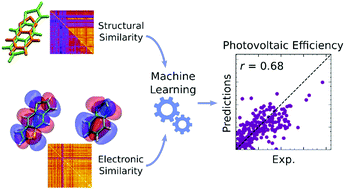
We present a translation of the chemical intuition in materials discovery, in terms of chemical similarity of efficient materials, into a rigorous framework exploiting machine learning. We computed equilibrium geometries and electronic properties (DFT) for a database of 249 Organic donor–acceptor pairs. We obtain similarity metrics between pairs of donors in terms of electronic and structural parameters, and we use such metrics to predict photovoltaic efficiency through linear and non-linear machine learning models. We observe that using only electronic or structural parameters leads to similar results, while considering both parameters at the same time improves the predictive capability of the models up to correlations of r ≈ 0.7. Such correlation allows for reliable predictions of efficient materials, and lends to be coupled with combinatorial of evolutionary approaches for a more reliable virtual screening of candidate materials.
- This article is part of the themed collection: Horizons Community Board Collection: Solar Energy Conversion
 Please wait while we load your content...
Please wait while we load your content...

