
Systematic design of analogs of active compounds covering more than 1000 targets†
Abstract
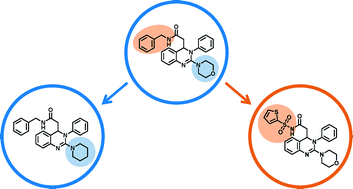
A computational methodology is presented for the systematic design of analogs. The approach is applicable to large data sets. Series of structurally related compounds are automatically detected, core structures and substituents isolated, novel core-substituent combinations enumerated, and synthetically accessible analogs identified. Compared to active compounds sharing the same core structure, virtual analogs often contain new substitution sites and chemical features. The methodology has been applied to compounds active against more than 1000 targets and has yielded nearly two million well-defined virtual analogs. A database comprising these analogs is made freely available as a resource for candidate selection and chemical exploration.
 Please wait while we load your content...
Please wait while we load your content...