
An improved approach for predicting drug–target interaction: proteochemometrics to molecular docking†
Abstract
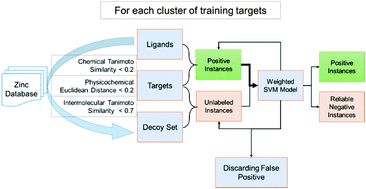
Proteochemometric (PCM) methods, which use descriptors of both the interacting species, i.e. drug and the target, are being successfully employed for the prediction of drug–target interactions (DTI). However, unavailability of non-interacting dataset and determining the applicability domain (AD) of model are a main concern in PCM modeling. In the present study, traditional PCM modeling was improved by devising novel methodologies for reliable negative dataset generation and fingerprint based AD analysis. In addition, various types of descriptors and classifiers were evaluated for their performance. The Random Forest and Support Vector Machine models outperformed the other classifiers (accuracies >98% and >89% for 10-fold cross validation and external validation, respectively). The type of protein descriptors had negligible effect on the developed models, encouraging the use of sequence-based descriptors over the structure-based descriptors. To establish the practical utility of built models, targets were predicted for approved anticancer drugs of natural origin. The molecular recognition interactions between the predicted drug–target pair were quantified with the help of a reverse molecular docking approach. The majority of predicted targets are known for anticancer therapy. These results thus correlate well with anticancer potential of the selected drugs. Interestingly, out of all predicted DTIs, thirty were found to be reported in the ChEMBL database, further validating the adopted methodology. The outcome of this study suggests that the proposed approach, involving use of the improved PCM methodology and molecular docking, can be successfully employed to elucidate the intricate mode of action for drug molecules as well as repositioning them for new therapeutic applications.
 Please wait while we load your content...
Please wait while we load your content...