
A network-based, integrative approach to identify genes with aberrant co-methylation in colorectal cancer†‡
Abstract
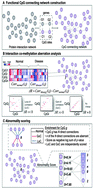
Epigenetic changes, including aberrations in DNA methylation, are a common hallmark of many cancers. The identification and interpretation of epigenetic changes associated with cancers may benefit from integration with protein interactomes. Based on the assumption that genes implicated in a specific tumor phenotype will show high aberrant co-methylation patterns with their interacting partners, we propose an integrated approach to uncover cancer-associated genes by integrating a DNA methylome with an interactome. Aberrant co-methylated interactions were first identified in the specific cancer, and genes were then prioritized based on their enrichment in aberrant co-methylation. By applying this to a large-scale colorectal cancer (CRC) dataset, the proposed method increases the power to capture known genes. More importantly, genes possessing high aberrant co-methylation patterns, located at the topological center of the original protein–protein interaction network (PPIN), affect several cancer-associated pathways and form hotspots that are frequently hijacked in cancer. Additionally, the top-ranked candidate genes may also be useful as an indicator of CRC diagnosis and prognosis. Five fold cross-validation of the top-ranked genes in diagnosis reveals that it can achieve an area under the receiver operating characteristic (ROC) curve ranging from 82.2% to 98.4% in three independent datasets. Five of these genes form a core repressive module. CCNA1 and ESR1 in particular are evidently silenced by promoter hypermethylation in CRC cell lines and tissues, whose re-expression markedly suppresses tumor cell survival and clonogenicity. These results show that the network-centric method could identify novel disease biomarkers and model how oncogenic lesions mediate epigenetic changes, providing important insights into tumorigenesis.
 Please wait while we load your content...
Please wait while we load your content...