
Towards measuring reactivity on micro-to-millisecond timescales with laser pump, NMR probe spectroscopy†

Abstract
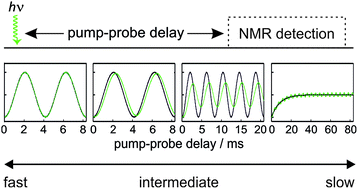
We present a quantitative analysis of the timescales of reactivity that are accessible to a laser pump, NMR probe spectroscopy method using para-hydrogen induced polarisation (PHIP) and identify three kinetic regimes: fast, intermediate and slow. These regimes are defined by the relative rate of reaction, k, compared to δω, the frequency of the NMR signal oscillations associated with the coherent evolution of the hyperpolarised 1H NMR signals created after para-hydrogen (p-H2) addition during the pump-probe delay. The kinetic regimes are quantitatively defined by a NMR dephasing parameter, ε = δω/k. For the fast regime, where k ≫ δω and ε tends to zero, the observed NMR signals are not affected by the chemical evolution of the system and so only an upper bound on k can be determined. In the slow regime, where k ≪ δω and ε tends to infinity, destructive interference leads to the complete dephasing of the coherent NMR signal intensity oscillations. As a result, the observed NMR signal evolution during the pump-probe delay reflects only the chemical change of the system and NMR relaxation. Finally, in the intermediate regime, where k ∼ δω, characteristic partial dephasing of the NMR signal oscillations is predicted. In the limit where the dephasing parameter is small but non-zero, chemical evolution manifests itself as a phase shift in the NMR signal oscillation that is equal to the dephasing parameter. As this phase shift is predicted to persist for pump-probe delays much longer than the timescale of the formation of the product molecules, it provides a route to measure reactivity on micro-to-millisecond timescales through NMR detection. We predict that the most significant fundamental limitations of the accessible reaction timescales are the duration of the NMR excitation pulse (∼1 μs) and the chemical shift difference (in Hz) between the p-H2-derived protons in the product molecule.
- This article is part of the themed collection: Mechanistic processes in organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

