
Modeling of internal conversion in photoexcited conjugated molecular donors used in organic photovoltaics†
Abstract
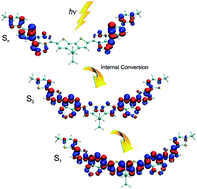
Using the Non-Adiabatic Excited States Molecular Dynamics (NA-ESMD) approach, we investigate the ultrafast electronic relaxation in a recently synthesized small molecule donor, p-DTS(PTTh2)2, which belongs to the dithienosilole-pyridylthiadiazole family of chromophores. In combination with the PC70BM acceptor, p-DTS(PTTh2)2 can be used to fabricate high efficiency bulk heterojunction organic solar cells. After photoexcitation to its broad high-energy peak in the 3–4 eV range, associated with multiple excited states, p-DTS(PTTh2)2 undergoes efficient ultrafast internal conversion to its lowest excited state. During this process, about 1–2 eV electronic energy transfers to the vibrational degrees of freedom leading to rapid heating of the molecule. Nevertheless, our simulations do not detect possible bond-breaking or decomposition of the system. This suggests minimal intra-molecular photodamage after photoexcitation to high-energy states in the 3–4 eV region. Calculated radiationless deactivation mainly consists of a sequential mechanism that involves electronic transitions between the current transient state and the corresponding state directly below in energy. Changes in the density of states along the relaxation process lead to pronounced variations and time-dependence of the accumulated populations of the different intermediate electronic excited states. Visualization of the electronic transition density during internal conversion reveals spatial intramolecular delocalization of electronic excitation from the thiophene moieties to the entire chromophore. Finally, our analysis of non-adiabatic coupling vectors suggests characteristic vibrational degrees of freedom coupled to the electronic system during various stages of non-radiative relaxation.
 Please wait while we load your content...
Please wait while we load your content...

