
Palladium(0) complexes of diferrocenylmercury diphosphines: synthesis, X-ray structure analyses, catalytic isomerization, and C–Cl bond activation†

Abstract
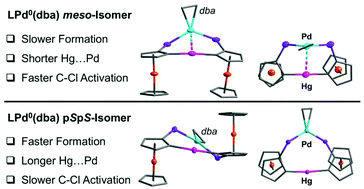
Palladium(0) phosphine complexes are of great importance as catalysts in numerous bond formation reactions that involve oxidative addition of substrates. Highly active catalysts with labile ligands are of particular interest but can be challenging to isolate and structurally characterize. We investigate here the synthesis and chemical reactivity of Pd0 complexes that contain geometrically adaptable diferrocenylmercury-bridged diphosphine chelate ligands (L) in combination with a labile dibenzylideneacetone (dba) ligand. The diastereomeric diphosphines 1a (pSpR, meso-isomer) and 1b (pSpS-isomer) differ in the orientation of the ferrocene moieties relative to the central Ph2PC5H3–Hg–C5H3PPh2 bridging entity. The structurally distinct trigonal LPd0(dba) complexes 2a (meso) and 2b (pSpS) are obtained upon treatment with Pd(dba)2. A competition reaction reveals that 1b reacts faster than 1a with Pd(dba)2. Unexpectedly, catalytic interconversion of 1a (meso) into 1b (rac) is observed at room temperature in the presence of only catalytic amounts of Pd(dba)2. Both Pd0 complexes, 2a and 2b, readily undergo oxidative addition into the C–Cl bond of CH2Cl2 at moderate temperatures with formation of the square-planar trans-chelate complexes LPdIICl(CH2Cl) (3a, 3b). Kinetic studies reveal a significantly higher reaction rate for the meso-isomer 2a in comparison to (pSpS)-2b.
 Please wait while we load your content...
Please wait while we load your content...

