
Controlling the lability of uranyl(vi) through intramolecular π–π stacking†
 bc
and
Koichiro
Takao
*a
bc
and
Koichiro
Takao
*a
Abstract
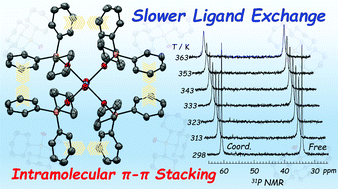
A reaction of UO22+ with cyclohexyldiphenylphosphine oxide (OPCyPh2) in ethanol resulted in a perchlorate salt of the 4-fold homoleptic complex, [UO2(OPCyPh2)4](ClO4)2·EtOH. X-ray structure determination revealed that [UO2(OPCyPh2)4]2+ shows a highly symmetric molecular structure supported by the intramolecular π–π stacking interactions between the phenyl groups of the neighbouring OPCyPh2 in the equatorial plane of UO22+. To clarify whether the reactivity of UO22+ is affected by such a unique coordination structure, the ligand exchange kinetics of [UO2(OPCyPh2)4]2+ in non-coordinating solvents has been studied using an NMR line-broadening method. In CD2Cl2 and CD3NO2 solutions, both signals of coordinated and free OPCyPh2 appeared distinctively even at 298 K, while 2D EXSY experiment provided evidence that a chemical exchange between [UO2(OPCyPh2)4]2+ and free OPCyPh2 occurs in these systems. Thus, this ligand exchange reaction is quite slow on the NMR time-scale. The dependency of the apparent first-order rate constant (kobs) on temperature and the free OPCyPh2 concentration suggested that this ligand exchange reaction involves associative and dissociative mechanisms both governed by large negative ΔS‡ terms predominantly. Compared with the kinetic data of ligand exchange reactions of other UO22+ complexes reported so far, the lability of UO22+ in [UO2(OPCyPh2)4]2+ is found to be significantly suppressed due to the intramolecular π–π stacking interactions as observed in the crystal structure. The DFT calculations successfully reproduced the intramolecular π–π stacking in [UO2(OPCyPh2)4]2+ by considering the empirical dispersion corrections, and provided theoretical rationales to the A and D mechanisms of the ligand exchange reaction of [UO2(OPCyPh2)4]2+.
 Please wait while we load your content...
Please wait while we load your content...

