
Molecular motions in a fluxional (η6-indenyl)tricarbonylchromium hemichelate: a density functional theory molecular dynamics study†

Abstract
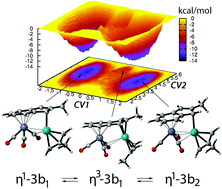
A comprehensive Density Functional Theory (DFT) study is reported on molecular motions occurring in a hemichelate complex, formally composed of the (η3-2-methylallyl)Pd(II) cation bonded to a (η6-indenyl)tricarbonylchromium anion, that has been recognized as being highly fluxional in a previous experimental study (C. Werlé, M. Hamdaoui, C. Bailly, X.-F. Le Goff, L. Brelot and J.-P. Djukic, J. Am. Chem. Soc., 2013, 135, 1715–1718). Its dynamics can be decomposed into three molecular motions: (i) the indenyl ligand can switch from a η1 to a η3 binding mode to Pd, (ii) the Cr(CO)3 moiety forms loose interactions with Pd, and can potentially rotate around the aryl-Cr axis, and (iii) the binding mode of the 2-methylallyl ligand to Pd may undergo partial decoordination or rotation around the Pd–C axis. Herein, DFT based molecular dynamics simulations have been performed to provide microscopic insights into the dynamical behaviour of the complex on a 100 ps timescale. QTAIM analyses have been performed over trajectories to investigate the rearrangement of its electronic structure over time. Next, free energy calculations, following the metadynamics approach, have been undertaken to compute thermodynamic and kinetic data relative to the molecular rearrangements occurring in the complex. The results show that the fastest motion is the ring slippage of the indenyl ligand, and that this motion is coupled with the rotation of the 2-methylallyl ligand. Conversely, the Cr(CO)3 rotation is significantly slower, however, a fast rocking motion is observed, consisting of transient approaches of carbonyl ligands to Pd. QTAIM analyses point to loose Pd/carbonyl and Pd/Cr interactions on average, that significantly evolve over time as fluxional motions occur.
 Please wait while we load your content...
Please wait while we load your content...

