
Ligand K-edge XAS, DFT, and TDDFT analysis of pincer linker variations in Rh(i) PNP complexes: reactivity insights from electronic structure†
Abstract
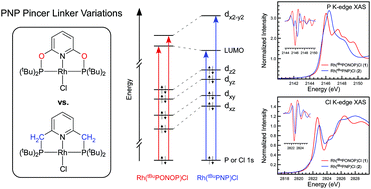
Here we report P K-edge, Cl K-edge, and Rh L3-edge X-ray absorption spectroscopy (XAS) data for Rh[C5H3N-2,6-(XPtBu2)2]Cl, where X = O (tBuPONOP; 1) or CH2 (tBuPNP; 2). Solid-state XAS data for 1 and 2 were compared to density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations to identify how changing the PNP pincer linker from O to CH2 affected electronic structure and bonding at Rh(I). Pronounced differences in XAS peak intensities and energies were observed. The P K-edge XAS data revealed a large increase in Rh 4dx2−y2 and P 3p orbital-mixing (Rh–P σ*) in 1 compared to 2, and pronounced transition energy variations reflected marked differences in orbital energies and compositions. By comparison, the Cl K-edge XAS data revealed only subtle differences in Rh–Cl covalency, although larger splitting between the Rh–Cl π* and σ* transitions was observed in 2. Analysis of the occupied MOs from DFT (HOMO, HOMO−1, HOMO−2, and HOMO−3) and comparison to the unoccupied MOs involved in XAS revealed a relatively uniform energy increase (ca. 0.3–0.5 eV) for all five 4d-derived molecular orbitals in Rh(tBuPNP)Cl (2) compared to Rh(tBuPONOP)Cl (1). The energy shift was relatively invariant with respect to differences in orbital symmetry, bonding type (σ or π), and orbital mixing, which suggested that the increase could be attributed to electrostatic effects. The change in d-orbital energies are consistent with known reactivity differences of Rh(tBuPONOP)+ and Rh(tBuPNP)+ towards CO, H2, and CH2Cl2, and are explained here by considering how d-orbital energies affect covalent L → M σ bonding and M → L π backbonding.
- This article is part of the themed collection: New Talent: Americas
 Please wait while we load your content...
Please wait while we load your content...

