
The strength of actinide–element bonds from the quantum theory of atoms-in-molecules†
Abstract
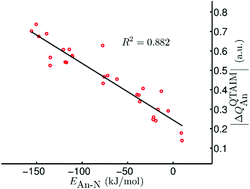
[AnX3]2(μ–η2:η2-N2) (An = Th–Pu; X = F, Cl, Br, Me, H, OPh) have been studied using relativistic density functional theory. Geometric and vibrational data suggest that metal→N2 charge transfer maximises at the protactinium systems, which feature the longest N–N bonds and the smallest σ(N–N), as a result of partial population of the N–N π* orbitals. There is very strong correlation of the standard quantum theory of atoms-in-molecules (QTAIM) metrics – bond critical point ρ, ∇2ρ and H and delocalisation indices – with An–N and N–N bond lengths and σ(N–N), but the correlation with An–N interaction energies is very poor. A similar situation exists for the other systems studied; neutral and cationic actinide monoxide and dioxides, and AnL3+ and AnL33+ (L = pyridine (Py), pyrazine (Pz) and triazine (Tz)) with the exception of some of the ∇2ρ data, for which moderate to good correlations with energy data are sometimes seen. By contrast, in almost all cases there is very strong correlation of interaction and bond energies with |ΔQQTAIMAn|, a simple QTAIM metric which measures the amount of charge transferred to or from the actinide on compound formation.
- This article is part of the themed collection: Dalton Discussion 14: Advancing the chemistry of the f-elements
 Please wait while we load your content...
Please wait while we load your content...

