
Electronic excited states of chromium and vanadium bisarene complexes revisited: interpretation of the absorption spectra on the basis of TD DFT calculations†
Abstract
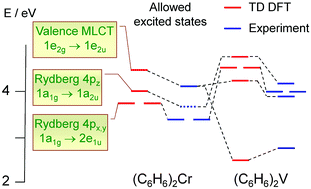
The nature and energies of the (arene)2Cr, (arene)2V and (arene)2Cr+ (arene = η6-C6H6, η6-C6H5Me, η6-1,3-C6H4Me2, and η6-1,3,5-C6H3Me3) electronic excited states have been determined on the basis of the time-dependent density functional theory (TD DFT) approach and comparison with the gas-phase and condensed-phase absorption spectra. Both valence-shell and Rydberg electronic excitations were taken into account. The TD DFT results appear to describe correctly the influence of the metal atom and the ligand on the band positions and intensities in the UV-visible absorption spectra as well as the mixing of Rydberg and intravalency states. The TD DFT calculations suggest new assignments for long-wavelength valence-shell absorption bands in the spectra of (arene)2V and (arene)2Cr+.
- This article is part of the themed collection: Spectroscopy of Inorganic Excited States
 Please wait while we load your content...
Please wait while we load your content...

