
Investigation of ternary ConCN−1/0 (n = 1–5) clusters by density functional calculations
Abstract
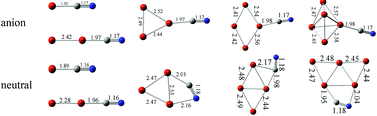
The neutral and anionic ConCN (n = 1–5) clusters were investigated using density functional calculations. The most stable structures of neutral and anionic ConCN (n = 1–5) clusters have been identified. In these structures, the CN radical retains its integrity as a structural unit. For anionic ConCN− (n = 1–5) and neutral ConCN (n = 1–2) clusters, the CN prefers to be adsorbed on the top sites of Con clusters via the C terminal, forming a linear or quasi-linear N–C–Co structure. For neutral ConCN (n = 3–5) clusters, the CN is adsorbed on the bridge sites of Con (n = 3–5) by both C and N atoms. Compared to the free CN radical calculated at the same level, the C–N stretching frequencies of neutral ConCN (n = 3–5) are red-shifted while they are blue-shifted for neutral ConCN (n = 1–2). The adiabatic and vertical detachment energies of anionic ConCN− (n = 1–5) clusters are calculated based on density functional calculations. In addition, the most favored dissociation channels of neutral and anionic ConCN (n = 1–5) clusters are determined by calculating the dissociation energies of various possible dissociation pathways.
 Please wait while we load your content...
Please wait while we load your content...

