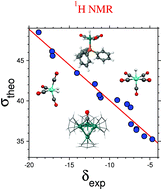
Transition metal hydrides are of great interest in chemistry because of their reactivity and their potential use as catalysts for hydrogenation. Among other available techniques, structural properties in transition metal (TM) complexes are often probed by NMR spectroscopy. In this paper we will show that it is possible to establish a viable methodological strategy in the context of density functional theory, that allows the determination of 1H NMR chemical shifts of hydride ligands attached to transition metal atoms in mononuclear systems and clusters with good accuracy with respect to experiment. 13C chemical shifts have also been considered in some cases. We have studied mononuclear ruthenium complexes such as Ru(L)(H)(dppm)2 with L = H or Cl, cationic complex [Ru(H)(H2O)(dppm)2]+ and Ru(H)2(dppm)(PPh3)2, in which hydride ligands are characterized by a negative 1H NMR chemical shift. For these complexes all calculations are in relatively good agreement compared to experimental data with errors not exceeding 20% except for the hydrogen atom in Ru(H)2(dppm)(PPh3)2. For this last complex, the relative error increases to 30%, probably owing to the necessity to take into account dynamical effects of phenyl groups. Carbonyl ligands are often encountered in coordination chemistry. Specific issues arise when calculating 1H or 13C NMR chemical shifts in TM carbonyl complexes. Indeed, while errors of 10 to 20% with respect to experiment are often considered good in the framework of density functional theory, this difference in the case of mononuclear carbonyl complexes culminates to 80%: results obtained with all-electron calculations are overall in very satisfactory agreement with experiment, the error in this case does not exceed 11% contrary to effective core potentials (ECPs) calculations which yield errors always larger than 20%. We conclude that for carbonyl groups the use of ECPs is not recommended, although their use could save time for very large systems, for instance in cluster chemistry. The reliance of NMR chemical shielding on dynamical effects, such as intramolecular rearrangements or trigonal twists, is also examined for H2Fe(CO)4, K+[HFe(CO)]−, HMn(CO)5 and HRe(CO)5. The accuracy of the theory is also examined for complexes with two dihydrogen ligands (Tp*RuH(H2)2 and [FeH(H2)(DMPE)2]+) and a ruthenium cluster, [H3Ru4(C6H6)4(CO)]+. It is shown that for all complexes studied in this work, the effect of the ligands on the chemical shielding of hydrogen coordinated to metal is suitably calculated, thus yielding a very good correlation between experimental chemical shifts and theoretical chemical shielding.

 Please wait while we load your content...
Please wait while we load your content...

