
Structure, equilibrium and ligand exchange dynamics in the binary and ternary dioxouranium(vi)-ethylenediamine-N,N′-diacetic acid-fluoride system: a potentiometric, NMR and X-ray crystallographic study†
Abstract
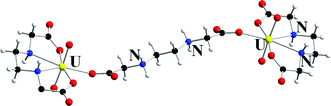
The structure, thermodynamics and kinetics of the binary and ternary uranium(VI)-ethylenediamine-N,N′-diacetate (in the following denoted EDDA) fluoride systems have been studied using potentiometry, 1H, 19F NMR spectroscopy and X-ray diffraction. The UO22+–EDDA system could be studied up to −log[H3O+] = 3.4 where the formation of two binary complexes UO2(EDDA)(aq) and UO2(H3EDDA)3+ were identified, with equilibrium constants logβ(UO2EDDA) = 11.63 ± 0.02 and logβ(UO2H3EDDA3+) = 1.77 ± 0.04, respectively. In the ternary system the complexes UO2(EDDA)F−, UO2(EDDA)(OH)− and (UO2)2(µ-OH)2(HEDDA)2F2(aq) were identified; the latter through 19F NMR. 1H NMR spectra indicate that the EDDA ligand is chelate bonded in UO2(EDDA)(aq), UO2(EDDA)F− and UO2(EDDA)(OH)− while only one carboxylate group is coordinated in UO2(H3EDDA)3+. The rate and mechanism of the fluoride exchange between UO2(EDDA)F− and free fluoride was studied by 19F NMR spectroscopy. Three reactions contribute to the exchange; (i) site exchange between UO2(EDDA)F− and free fluoride without any net chemical exchange, (ii) replacement of the coordinated fluoride with OH− and (iii) the self dissociation of the coordinated fluoride forming UO2(EDDA)(aq); these reactions seem to follow associative mechanisms. 1H NMR spectra show that the exchange between the free and chelate bonded EDDA is slow and consists of several steps, protonation/deprotonation and chelate ring opening/ring closure, the mechanism cannot be elucidated from the available data. The structure (UO2)2(EDDA)2(µ-H2EDDA) was determined by single crystal X-ray diffraction and contains two UO2(EDDA) units with tetracoordinated EDDA linked by H2EDDA in the “zwitterion” form, coordinated through a single carboxylate oxygen from each end to the two uranium atoms. The geometry of the complexes indicates that there is no geometric constraint for an associative ligand substitution mechanism.
 Please wait while we load your content...
Please wait while we load your content...

