
Identifying the roles of acid–base sites in formation pathways of tolualdehydes from acetaldehyde over MgO-based catalysts†

Abstract
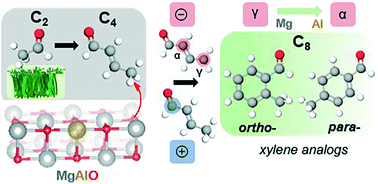
Pure and Al-substituted MgO catalysts are studied to identify the contributions of acid–base sites in the formation of two valuable xylene analogs, ortho- and para-tolualdehydes, from an ethanol derivative, acetaldehyde. The catalyst properties are characterized through XRD, 27Al MAS NMR, ICP-AES, N2 physisorption, TPD-MS, and DRIFTS experiments. Reactivity comparisons of untreated and CO2-titrated catalysts at 250 °C, coupled with CO2 DRIFTS studies on fresh and spent samples, indicate the formation of tolualdehydes from intermediates is initiated through deprotonation by a medium-strength basic site in a specific, metal–oxygen (M–O)-type coordination environment. Analyses of the catalytic surface properties and reactivity, pathways of formation, and natural bond orbital (NBO) charge distribution suggest C4 + C4 (rather than C2 + C6) mechanistic steps dominate tolualdehyde production over these catalysts under the investigated reaction conditions. Isomeric selectivity to ortho-tolualdehyde is 92 and 81 mol% over pure and Al-substituted MgO catalysts, respectively. We propose that the shift in isomeric selectivity towards para- upon introduction of a proximal Lewis acidic functionality (Al3+/MgO) to the catalyst is caused by electron redistribution in the conjugated enolate from the γ-C (forming ortho-) towards the α-C (forming para-) due to the carbonyl-O/Lewis acid coordination. This insight provides a framework for the development of next generation catalysts that give improved reactivity in cascade reactions of C2 feedstocks to aromatics.
 Please wait while we load your content...
Please wait while we load your content...

