
Toward rational catalyst design for partial hydrogenation of dimethyl oxalate to methyl glycolate: a descriptor-based microkinetic analysis†

Abstract
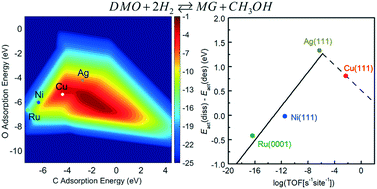
Partial hydrogenation of dimethyl oxalate (DMO) has attained increasing attention in the chemical industry, which is driven by the desire to develop a means of achieving methyl glycolate (MG) production in a more economical and eco-friendly way than the traditional petroleum route. In this contribution, a descriptor-based microkinetic analysis combined with results from density functional theory calculations has been performed to examine the reaction mechanism for DMO hydrogenation to MG on Cu, Ag, Ni, and Ru catalysts. Calculated results indicate that, along the dominant reaction pathway, DMO molecules are first dissociated into methoxyl and acyl species on the catalyst surfaces, followed by the hydrogenation of methoxyl and the successive hydrogenation of acyl at the carbon and oxygen atoms of the terminal carbonyl group. Linear chemisorption and transition state energy scaling relations indicate that the adsorption energies of atomic C and O can be used as descriptors to represent the energetics of other adsorbed species and transition states. Both energy profiles and a volcano-shaped activity map indicate that among the four catalysts Cu is the most effective in the hydrogenation reaction, and the activity varies in the order Cu > Ag > Ni > Ru. The catalyst selectivity is found to initially increase with the catalytic activity and then decline, also giving a volcano curve. The reason Ag has a better selectivity than Cu lies in the fact that the greater binding strength of adsorbates to the Cu surface not only promotes DMO dissociation but also hinders the desorption of MG.
 Please wait while we load your content...
Please wait while we load your content...

