
Mechanism of NO–CO reaction over highly dispersed cuprous oxide on γ-alumina catalyst using a metal–support interfacial site in the presence of oxygen: similarities to and differences from biological systems†
 *ab
Masahiro
Ehara,
ac
Saburo
Hosokawa,
ab
Tsunehiro
Tanaka
ab
and
Shigeyoshi
Sakaki
*ad
*ab
Masahiro
Ehara,
ac
Saburo
Hosokawa,
ab
Tsunehiro
Tanaka
ab
and
Shigeyoshi
Sakaki
*ad
Abstract
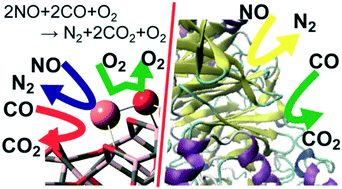
Copper–alumina (Cu/Al2O3) systems exhibit highly catalytic activity for nitric oxide reduction with carbon monoxide (NO–CO reaction) even in the presence of dioxygen molecule, but the origin of the interesting catalysis remains unclear. Herein, we elucidated the NO–CO reaction mechanism over the Cu/γ-Al2O3 catalyst using DFT and a cluster model consisting of a single Cu2O unit loaded on to a γ-Al2O3 cluster. The DFT calculations showed that the reactions occur via the Cu+/Cu2+ catalytic cycle, which starts from NO dimerization, followed by N2O formation via the first N–O bond scission. The next step is the rate-determining N2O decomposition with the simultaneous formation of a Cu2+ site. The resultant Cu2+ site weakly adsorbs a CO molecule. CO oxidation by surface oxygen occurs with a small activation barrier. On the contrary, CO oxidation with molecular O2 over Cu/γ-Al2O3 is kinetically unfavourable because it needs a large activation energy. The Cu–Al interface plays a crucially important role in NO dimerization and N2O decomposition, which indicates the importance of highly dispersed Cu over Al2O3. Notably, the highly dispersed Cu is not easily oxidized by O2 because O–O bond cleavage is unfavourable compared to N2–O bond cleavage over Cu/γ-Al2O3. This is the origin of the high catalytic activity of the Cu/γ-Al2O3 for the NO–CO reaction even in the presence of O2. The characteristic features of this reaction are similar to NO reduction by nitric oxide/nitrous oxide reductases and CO oxidation by molybdenum CO dehydrogenase from the viewpoint of the key intermediates and electronic processes of the catalytic reaction.
 Please wait while we load your content...
Please wait while we load your content...

