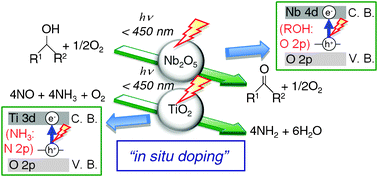
This paper reviews the recent development of the photocatalytic emission control reaction and photooxidation with molecular oxygen, specifically focusing on efforts based on revealing the reaction mechanism by the authors' group. TiO2 acts as an effective catalyst for the photo-assisted selective catalytic reduction of NO with NH3 in the presence of O2 (photo-SCR). Photooxidation of alcohols to carbonyl compounds proceeds selectively over Nb2O5 without organic solvents. Usually, both TiO2 and Nb2O5 work only in the ultraviolet (UV) region because of the limit of their bandgap energies. However, both photo-SCR over TiO2 and photooxidation of alcohols over Nb2O5 proceed even under visible light irradiation up to ca. 450 nm. This indicates that these two reactions take place by the different photo-activation mechanism from the classical electron transfer mechanism in semiconductor photocatalysis, that is, the formation of an excited electron in the conduction band and the positive hole in the valence band. A mechanistic study using UV-Vis, ESR, FT/IR, kinetic study, and DFT calculations revealed the reaction mechanisms of photo-SCR and photooxidation of alcohols, and that the surface complex consisting of the adsorbed molecule and catalyst plays an important role in the photo-activation step. The surface complex is converted to the photo-activated species even under visible light irradiation, because the direct electron transition from a donor level derived from the adsorbed molecule to the conduction band of a photocatalyst takes place and a photo-generated hole is trapped on the adsorbed molecule to form the photo-activated radical species. The effective wavelength is shifted to a longer wavelength by the formation of the donor level derived from the adsorbed molecule during a chemical reaction (called here “in situ doping”). This unique photo-activation mechanism by “in situ doping” gives us attractive ways for removing the limit of bandgap energy, and the utilization of visible light.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


