
Peptide macrocyclization by transition metal catalysis
 *ab
Gerardo M.
Ojeda-Carralero,
ab
Leslie
Reguera
b
and
Erik V.
Van der Eycken
*ac
*ab
Gerardo M.
Ojeda-Carralero,
ab
Leslie
Reguera
b
and
Erik V.
Van der Eycken
*ac
Abstract
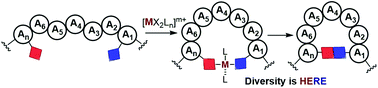
Peptide macrocyclization has traditionally relied on lactam, lactone and disulfide bond-forming reactions that aim at introducing conformational constraints into small peptide sequences. With the advent of ruthenium-catalyzed ring-closing metathesis and copper-catalyzed alkyne–azide cycloaddition, peptide chemists embraced transition metal catalysis as a powerful macrocyclization tool with relevant applications in chemical biological and peptide drug discovery. This article provides a comprehensive overview of the reactivity and methodological diversification of metal-catalyzed peptide macrocyclization as a special class of late-stage peptide derivatization method. We report the evolution from classic palladium-catalyzed cross-coupling approaches to more modern oxidative versions based on C–H activation, heteroatom alkylation/arylation and annulation processes, in which aspects such as chemoselectivity and diversity generation at the ring-closing moiety became dominant over the last years. The transit from early cycloadditions and alkyne couplings as ring-closing steps to very recent 3d metal-catalyzed macrocyclization methods is highlighted. Similarly, the new trends in decarboxylative radical macrocyclizations and the interplay between photoredox and transition metal catalysis are included. This review charts future perspectives in the field hoping to encourage further progress and applications, while bringing attention to the countless possibilities available by diversifying not only the metal, but also the reactivity modes and tactics to bring peptide functional groups together and produce structurally diverse macrocycles.
- This article is part of the themed collection: Celebrating Latin American Talent in Chemistry
 Please wait while we load your content...
Please wait while we load your content...

