
Towards operando computational modeling in heterogeneous catalysis
 a
Christopher J.
Heard,
a
Mikhail V.
Polynski,
b
Jittima
Meeprasert,
c
Evgeny A.
Pidko
*bc
and
Petr
Nachtigall
*a
a
Christopher J.
Heard,
a
Mikhail V.
Polynski,
b
Jittima
Meeprasert,
c
Evgeny A.
Pidko
*bc
and
Petr
Nachtigall
*a
Abstract
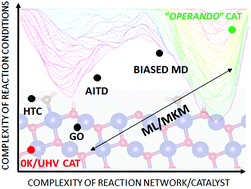
An increased synergy between experimental and theoretical investigations in heterogeneous catalysis has become apparent during the last decade. Experimental work has extended from ultra-high vacuum and low temperature towards operando conditions. These developments have motivated the computational community to move from standard descriptive computational models, based on inspection of the potential energy surface at 0 K and low reactant concentrations (0 K/UHV model), to more realistic conditions. The transition from 0 K/UHV to operando models has been backed by significant developments in computer hardware and software over the past few decades. New methodological developments, designed to overcome part of the gap between 0 K/UHV and operando conditions, include (i) global optimization techniques, (ii) ab initio constrained thermodynamics, (iii) biased molecular dynamics, (iv) microkinetic models of reaction networks and (v) machine learning approaches. The importance of the transition is highlighted by discussing how the molecular level picture of catalytic sites and the associated reaction mechanisms changes when the chemical environment, pressure and temperature effects are correctly accounted for in molecular simulations. It is the purpose of this review to discuss each method on an equal footing, and to draw connections between methods, particularly where they may be applied in combination.
- This article is part of the themed collections: In memory of Petr Nachtigall and New catalytic materials for energy and chemistry in transition
 Please wait while we load your content...
Please wait while we load your content...

