
The stability of coordination polyhedrons and distribution of europium ions in Ca6BaP4O17†
 a
Hongbin
Liang
*a
a
Hongbin
Liang
*a
Abstract
In this work, the coordination polyhedron stabilities and distributions of europium ions in Ca6BaP4O17 (CBPO) luminescent materials are investigated. The density functional theory (DFT)-based first principles calculation results show that the PO4 tetrahedrons can tilt in the structure, which leads to the atomic distortion of O13 and O12 in CBPO and the Eu2+/Eu3+-doped systems. The energy scale of about ∼0.1 eV suggests that stabilities of coordination polyhedrons are easily influenced by dynamic factors. The atomic distortion and vacancy of 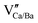 work as charge compensations in CBPO:Eu3+, and three lattice sites of europium are extracted and summarized. The X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) confirm that Eu3+ can occupy the Ca1, Ca2 and Ba sites of CBPO. The combination of first principles calculation and X-ray absorption fine structure (XAFS) provides more information about microstructures of luminescent materials.
work as charge compensations in CBPO:Eu3+, and three lattice sites of europium are extracted and summarized. The X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) confirm that Eu3+ can occupy the Ca1, Ca2 and Ba sites of CBPO. The combination of first principles calculation and X-ray absorption fine structure (XAFS) provides more information about microstructures of luminescent materials.
 Please wait while we load your content...
Please wait while we load your content...

