
On the elaboration of the next generation of thermodynamic models of solid solutions†

Abstract
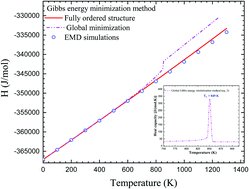
Thermodynamic models of solid solutions used in computational thermochemistry have not been modernized in recent years. With the advent of fast and cheap computers, it is nowadays possible to add, at a minimal computational cost, physical ingredients such as coordination numbers, inter-atomic distances and classical interatomic potentials to the function describing the energetics of ordered and disordered solid solutions. As we show here, the integration of these elements into a robust statistical thermodynamic model of solution establishes natural connections with other deterministic and stochastic atomistic methods such as Monte Carlo and molecular dynamics simulations. Ultimately, all these numerical approaches need to be self-consistent and generate complementary sets of numerical thermo-physical properties. The present work proposes a new formalism to define the Gibbs free energy of ordered and disordered solid solutions. It allows for a complete prediction of the thermal, volumetric and compositional dependence of the Gibbs free energy by solving a constrained minimization problem. As a proof of concept, we explore the energetic behavior of pure face-centered cubic gold as well as the AuCu L10 ordered solution as a function of both temperature and pressure. We finally compare these results with the average properties obtained from classical molecular dynamics simulations and explain the origin of the existing differences between the two approaches based on how the temperature is accounted for in each method.
 Please wait while we load your content...
Please wait while we load your content...

