
On the mechanism of predominant urea formation from thermal degradation of CO2-loaded aqueous ethylenediamine†

Abstract
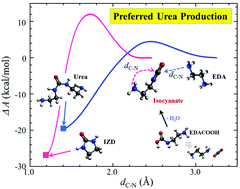
This study attempts to explain the well-known experimental observation that 1,3-bis(2-aminoethyl)urea (urea) is preferentially formed over the other major product, 2-imidazolidone (IZD), from thermal degradation of aqueous ethylenediamine (EDA) during the CO2 capture process. This is in direct contrast to the case of monoethanolamine (MEA), preferentially forming oxazolidinone (OZD), rather than urea, which undergoes further reactions leading to more stable products. Given their similar molecular structures, the different preferred degradation pathways of EDA and MEA impose an intriguing question regarding the underlying mechanism responsible for the distinct difference. Thermal degradation of both EDA and MEA tends to proceed mainly via formation of an isocyanate intermediate that may further undergo either cyclization to IZD (or OZD) or a reaction with EDA (or MEA) forming urea. For the EDA case, our first-principles simulations clearly demonstrate that the urea formation reaction is kinetically more, but thermodynamically less, favorable than the cyclization reaction; the opposite is true for the MEA case. Our further analysis shows that EDA–isocyanate is less prone to cyclization than MEA–isocyanate, which in turn allows for more facile urea formation. The reconfiguration dynamics of isocyanate is found to be governed by the dynamic nature of the interaction between its terminal group and surrounding solvent molecules. Our work highlights the importance of kinetic effects associated with the local structure and dynamics of solvent molecules around the intermediates that may significantly alter the degradation process of amine solvents.
 Please wait while we load your content...
Please wait while we load your content...

