
Ab initio investigation of the formation mechanism of nano-interfaces between 3d-late transition-metals and ZrO2 nanoclusters†

Abstract
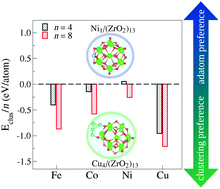
Understanding the formation of nano-interfaces between metallic clusters and nanoscale metal-oxides is an important step towards using such systems for catalytic applications. Thus, in this work, we employ density functional theory calculations to study the TMn–(ZrO2)13 interactions, for TM = Fe, Co, Ni, or Cu, and n = 1, 4, and 8. We found a general trend for adsorption and interaction energies (ad/int) for all cluster sizes, with 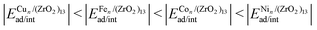 . In terms of size effects, both adsorption and interaction (frozen adsorbed structures) energies become stronger with increasing cluster sizes due to the increase in the number of TM atoms in direct contact with the (ZrO2)13 nanocluster. The structural and electronic properties change for each TMn/(ZrO2)13 system, indicating that these properties could be tuned through variables like the TM species, cluster size and morphology (isomers with different structures). The results also indicate that, from the studied TMs, Ni (Cu) should form the smallest (largest) clusters when supported on the (ZrO2)13 nanoclusters. These and other results discussed here help understand the formation of the nano-interface in the TM–ZrO2 systems, which can be useful in the development of new catalysts.
. In terms of size effects, both adsorption and interaction (frozen adsorbed structures) energies become stronger with increasing cluster sizes due to the increase in the number of TM atoms in direct contact with the (ZrO2)13 nanocluster. The structural and electronic properties change for each TMn/(ZrO2)13 system, indicating that these properties could be tuned through variables like the TM species, cluster size and morphology (isomers with different structures). The results also indicate that, from the studied TMs, Ni (Cu) should form the smallest (largest) clusters when supported on the (ZrO2)13 nanoclusters. These and other results discussed here help understand the formation of the nano-interface in the TM–ZrO2 systems, which can be useful in the development of new catalysts.
 Please wait while we load your content...
Please wait while we load your content...

