
Theoretical free energy profile and benchmarking of functionals for amino-thiourea organocatalyzed nitro-Michael addition reaction†

Abstract
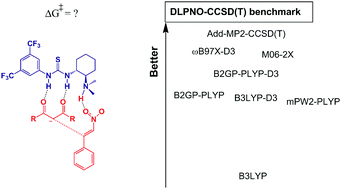
Amino-thiourea organocatalysis is an important catalytic process for enantioselective conjugate addition reactions. The interaction of the reactants with the catalyst has a substantial effect of dispersion forces and is a challenge for a reliable description when applying density functional theory. In this report, the classical addition of acetylacetone to β-nitro-styrene catalyzed by Takemoto's catalyst in toluene was studied using the PBE functional for geometry optimization and the DLPNO-CCSD(T) benchmark method for single point energy. The complete free energy profile calculated for the reaction was able to explain all experimental observations, including the fact that the carbon–carbon bond formation step is rate-determining. The overall barrier was calculated to be 22.8 kcal mol−1 (experimental value approximately 20 kcal mol−1), and the enantiomeric excess was calculated to be 88% (experimental value in the range of 84 to 92%). Some functionals were tested for single point energy. The hybrid B3LYP presented a high mean absolute deviation (MAD) from the DLPNO-CCSD(T) benchmark method by approximately 20 kcal mol−1. The inclusion of empirical dispersion correction in the B3LYP method decreased the MAD to 6 kcal mol−1. Even the double-hybrid mPW2-PLYP and B2GP-PLYP methods had MAD values of approximately 5 kcal mol−1. The inclusion of the dispersion correction decreased the MAD to 3.6 kcal mol−1. M06-2X and ωB97X-D3 were the most accurate among the tested functionals, with MADs of 2.5 kcal mol−1 and 1.8 kcal mol−1, respectively. Additivity approximation of the correlation energy was also tested and presented a MAD of only 0.6 kcal mol−1.
 Please wait while we load your content...
Please wait while we load your content...

