
Ultrafast dynamics in LMCT and intraconfigurational excited states in hexahaloiridates(iv), models for heavy transition metal complexes and building blocks of quantum correlated materials†

Abstract
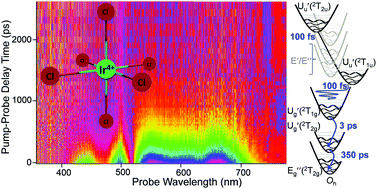
The population and structural dynamics of IrCl62− is studied in acetonitrile and aqueous solutions in comparison to isoelectronic IrBr62− using ultrafast broadband, dispersed transient absorption, with both octahedra excited with 85 fs pulses at four different wavelengths, encompassing the first seven t2g-based electronic states. Ligand-to-metal charge transfer (LMCT) 420 or 490 nm excitation of IrCl62− into Uu′(2T2u) + Eu′′(2T2u) states, superimposed due to Ham effect, or Uu′(2T1u), respectively, leads to symmetry lowering due to Jahn–Teller effect in these excited states with the subsequent 100 fs decay into Ug′(2T1g). This first LMCT state is formed vibrationally coherent in the 104 cm−1 t2g (scissor) or 243 cm−1 eg (out-of-phase-stretch) Jahn–Teller modes for the respective excitation wavelength. Direct excitation into Ug′(2T1g) at 600 nm and the intraconfigurational lowest excited Ug′(2T2g) state at 1900 nm helped to establish that Ug′(2T1g) decays via back electron transfer into Ug′(2T2g) (time constants, 3.55 ps in acetonitrile and 0.9 ps in water), and the decay of Ug′(2T2g) into the ground state is the rate-limiting relaxation step. The relaxation cascade of IrBr62− is similar with short-lived (≤100 fs) higher LMCT states, but the vibrational coherence is only observed in the Jahn–Teller t2g mode. Faster back electron transfer for IrBr62− is explained by the energy gap law. The intraconfigurational Ug′(2T2g) states, which are ∼5100 cm−1 above the ground state for both complexes, have a sub-nanosecond lifetime largely independent of the ligand nature (∼350 ps, acetonitrile).
 Please wait while we load your content...
Please wait while we load your content...

