
A global ab initio potential energy surface and dynamics of the proton-transfer reaction: OH− + D2 → HOD + D−†
 *b
Xueming
Yang
ab
and
*b
Xueming
Yang
ab
and
Abstract
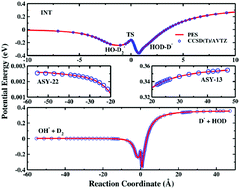
We report an accurate full-dimensional potential energy surface (PES) of the anion–molecule system OH3−. The PES was constructed by fitting 55 406 ab inito energies from the CCSD(T)/aug-cc-pVTZ level of theory with the fundamental invariant neural network (FI-NN) approach, resulting in an extremely small fitting error of 0.52 meV. Extensive quasiclassical trajectory (QCT) simulations were carried out on the PES to investigate the proton transfer dynamics (OH− + D2 → D− + HOD). The product D− translational energy distribution and angular distribution were calculated and compared with previous experimental measurements, in which reasonably good agreement has been achieved. The angular distribution at a high collision energy exhibits an exclusively forward scattering peak, indicating the direct stripping mechanism at high energies. With the decrease of the collision energy, the reaction shows a predominantly forward scattering feature, with very small sideways and backward scattering amplitudes, revealing combined mechanisms from direct abstraction with a short reaction time and a complex-forming process with a long reaction time.
- This article is part of the themed collection: Emerging AI Approaches in Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

