
On the influence of exact exchange on transition metal superatoms†
 a
and
N.
Gaston
*a
a
and
N.
Gaston
*a
Abstract
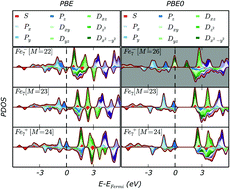
The electronic structure of A7C (A = Hg, Pd, V, Cr, Mn, Fe, Ni, Cu; C = 0, ±1, ±2) clusters has been determined using density functional theory methods. The A7C (A = Hg, Pd, Cr, Cu; C = 0, ±1, ±2) clusters all conform to the existing superatomic model, with a sufficiently stabilised local structure to prevent perturbation upon the introduction of exact exchange to the exchange correlation functional. For the A7C (A = Mn, Fe, Ni; C = 0, ±1, ±2) clusters the incorporation of exact exchange separates the atomic s- and d-electrons, leading to a net increase in the number of superatomic electrons. Conversely the incorporation of exact exchange into the exchange correlation functional decreases the number of superatomic electrons for the V7C (C = 0, ±1, ±2) clusters, owing to the radial extension of the d-orbitals influencing their ability to contribute into superatomic shells.
 Please wait while we load your content...
Please wait while we load your content...

