
First-principles study of phase stability, electronic and mechanical properties of plutonium sub-oxides†
 *a
and
*a
and
Abstract
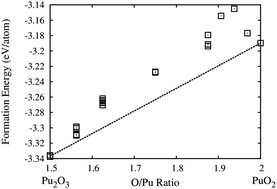
The formation energies (ΔHf) of fluorite PuO2, α-Pu2O3 and sub-oxides PuO2−x (0.0 < x < 0.5) are determined from atomic scale simulations based on density functional theory (DFT) employing the generalised gradient approximation (GGA) corrected with an effective Hubbard parameter (Ueff). The variation of structural and electronic properties of PuO2 and α-Pu2O3 is determined while ramping up Ueff from 0 eV to 5 eV (Ueff-ramping method) to treat the presence of metastable magnetic states and to determine the most suitable Ueff value matching the experiments. The GGA+U calculated lattice parameter variation as a function of stoichiometry (a(x)) for PuO2−x shows a positive volume of relaxation and an almost linear variation presented by the relation a(x) = a0 − 0.522738x, where a0 is the equilibrium lattice parameter of PuO2. The GGA+U calculated ΔHf values of PuO2−x lie above the tie line connecting the ΔHf of PuO2 and Pu2O3, and with decreasing O/Pu ratio, the stability of the sub-oxides increases. The crystal and electronic structure analysis of the oxygen vacancy in PuO2 shows outward anisotropic relaxation of four Pu atoms around the vacancy site. The electronic charges within the Wigner–Seitz sphere around these Pu atoms show an overall gain of only (0.12–0.22)e per Pu atom, signifying an incomplete localization of charges. Finally, the GGA+U calculated single crystal elastic constant values decrease continuously with decreasing O/Pu ratio from 2.0 to 1.5. The rate of decrease of the average C11 is almost 11–15 times higher compared to the rate of decrease of C12 and C44.
 Please wait while we load your content...
Please wait while we load your content...

