
Electronic structure and localized states in amorphous Si and hydrogenated amorphous Si†

Abstract
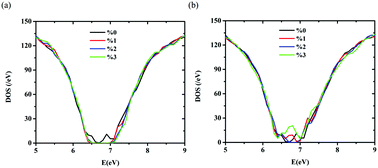
Hydrogen incorporation in the fabrication of amorphous Si (a-Si) plays an important role in improving its electronic and optical properties. An important question is how H interacts with the a-Si atomic network, and consequently affects the electronic properties of a-Si. The common assumption is that the role of H is to passivate the dangling bonds (DBs) of the a-Si structure, which subsequently leads to a reduction in the density of midgap sates and localized states within the mobility gap. In the present work, we first employ a combined molecular dynamic (MD) and density functional theory (DFT) method to create stable configurations of a-Si:H, and then analyze the atomic and electronic structure to investigate which structural defects interact with H, and how the electronic structure changes with H addition. We show that in contrast with the simple dangling bond picture, atoms bonded by highly strained bonds (SBs) are significantly affected by the addition of H, in terms of the lowest energy configuration, with similar if not greater importance to that of dangling bonds in passivating a-Si. We find that H atoms decrease the density of mid-gap states of a-Si by bonding to the Si atoms with SBs. Our results also indicate that Si atoms with SBs creates highly localized orbitals in the mobility gap of a-Si and a-Si:H, and the bonding of H atoms to them can significantly decrease the degree of orbital localization. The results demonstrate the beneficial effects of hydrogenation of a-Si in terms of reducing the overall strain energy of the a-Si network, with commensurate reduction of mid-gap states and orbital localization.
 Please wait while we load your content...
Please wait while we load your content...

