
Effect of hindered internal rotation treatments on predicting the thermodynamic properties of alkanes†
 *ab
*ab
Abstract
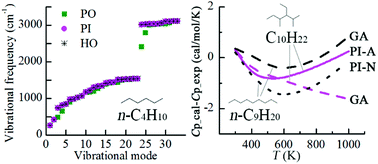
When considering hindered internal rotation, we usually have several options, including (i) single structure harmonic oscillator (SS-HO) approximation that considers the lowest-energy conformer only and approximates all molecular vibrations as harmonic oscillations, (ii) one-dimensional (1-D) internal rotation treatment that replaces the corresponding vibrational mode with one-dimensional torsion, and (iii) the multistructural method with torsional anharmonicity (MS-T) that considers the multiple-structure and torsional anharmonicity. These methods differ greatly in computational cost and accuracy. To evaluate the effect of different treatments on predicting thermodynamic properties, we calculated enthalpy, entropy, and heat capacity for a series of normal and branched alkanes using six different methods, including the SS-HO treatment, three 1-D methods, the MS-T method, and the group additivity (GA) method. The comparison of the computational results with experimental data shows that GA and two 1-D methods proposed in this study are more suitable for reliable and rapid predictions of thermodynamic properties for large hydrocarbons with many carbon–carbon single bonds.
 Please wait while we load your content...
Please wait while we load your content...

