
A computational study on ligand assisted vs. ligand participation mechanisms for CO2 hydrogenation: importance of bifunctional ligand based catalysts†

Abstract
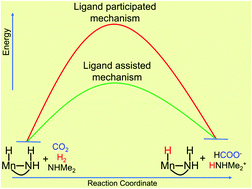
CO2 hydrogenation products are not only useful chemical sources but also promising hydrogen storage materials. A DFT study has been carried out on the CO2 hydrogenation reaction catalyzed by a series of bifunctional aminomethyl based Mn(I) complexes. We find that the N–H functionality in the aminomethyl ligand shows a metal–ligand cooperation (MLC) mechanism for the CO2 hydrogenation reaction. Here, the N–H functionality assists the MLC mechanism by stabilizing the formate anion via N–H⋯O hydrogen bonding interactions. This is opposite to the MLC mechanism proposed by Noyori for ketone hydrogenation, where the N–H functionality actively participates in the reaction mechanism via cleavage/formation of N–H/M–H bonds. Furthermore, the stabilized formate anion initiates heterolytic H2 cleavage, which requires a very low barrier compared to external base/ligand participation heterolytic H2 cleavage. Therefore, the bifunctional aminomethyl based Mn(I) complexes are promising for the CO2 hydrogenation reaction and our study may be very helpful for experimentalists for the development of efficient bifunctional ligand-based catalysts for the CO2 hydrogenation reaction.
 Please wait while we load your content...
Please wait while we load your content...

