
Effects of biaxial tensile strain on the first-principles-driven thermal conductivity of buckled arsenene and phosphorene†
 *a
*a
Abstract
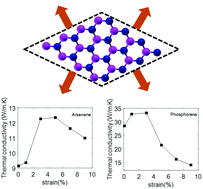
Strain engineering is an effective way to tune the thermal and electrical properties of novel two-dimensional (2D) materials. In this work, first-principles density functional theory (DFT) is used to systematically investigate the strain-dependent lattice thermal conductivity and phonon properties of buckled arsenene and phosphorene, which are the 2D materials with the highest thermal conductivities among monolayers in group-VA. We implemented an iterative self-consistent solution to the Peierls–Boltzmann transport equation (PBTE). Our results showed that the thermal conductivity in both monolayers exhibits an up-and-down behavior when biaxial tensile strain is applied in the range from 0% to 9%. The peak values in the thermal conductivities occur at 5% of strain in arsenene and 3% in phosphorene, with the maximum conductivities of strained arsenene and phosphorene being 1.4 and 1.2 times higher than those of unstrained samples, respectively. We provide a rigorous description of the underlying phonon physics responsible for these thermal responses to strain, addressing the interplay between phonon group velocities, heat capacities, and relaxation times. The acoustic-optical phonon band gaps in arsenene and phosphorene were found to reduce with strain, being the reduction more significant in phosphorene. Our results also showed that the use of the single mode relaxation time approximation (SMRTA) predict substantially lower thermal conductivities for arsenene and phosphorene than those predicted by the iterative solution of the PBTE.
 Please wait while we load your content...
Please wait while we load your content...

