
Assignment of photoelectron spectra of intramolecular silicon complexes: 1-vinyl- and 1-phenylsilatranes†

Abstract
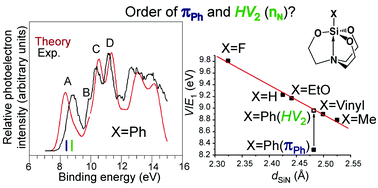
The problematic experimental photoelectron spectra of silatranes XSi[OCH2CH2]3N (X = vinyl and Ph) were theoretically studied. The ab initio electron propagator theory, many-body methods, and model vibrational Hamiltonian were employed to establish the nature of bands in the energetically lowest part of the photoelectron spectrum of these intramolecular silicon complexes. The first vertical ionization energy corresponds to a nitrogen atom lone pair, nN, in 1-vinylsilatrane, and the essentially doubly degenerate π-orbitals πPh and πPh′ of the aromatic ring in 1-phenylsilatrane. Peaks at 9.6 and 9.9 eV in the spectra of both compounds, previously associated with the removal of an electron from the lone pair level of the equatorial oxygens, should actually be assigned to the bonding orbital HV1 of their 3c–4e axial CSi←N moiety. Taking silatranes with X = H, Me, OEt, F, vinyl, and Ph as examples, it was found that the length of the dative Si←N contact and the ionization energy of HV2 (formally nN) were in a good linear relationship. Regardless of the nature of the X substituent, this relationship could be used for the identification of orbitals that participate in the formation of the coordination Si←N bond in the wide series of XSi[OCH2CH2]3N and their structural analogs.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

