
Excited state intramolecular proton transfer in julolidine derivatives: an ab initio study†
 a
and
Denis
Jacquemin
*b
a
and
Denis
Jacquemin
*b
Abstract
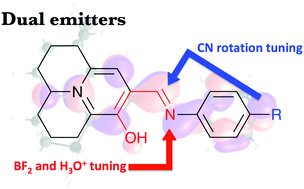
We have studied, using ab initio tools, a series of recently prepared fluorescent julolidine derivatives, undergoing Excited State Intramolecular Proton Transfer (ESIPT). We show that the computed free energy change in the excited state (ΔGES) can be used to predict the preference for enol, keto, or dual emission. Indeed, two julolidine molecules experimentally show dual emission, consistent with our finding of a small ΔGES. In agreement with experimental outcomes the complexation between the ESIPT centre and BF2 increases the rigidity of the fluorophore and greatly facilitates emission at energies close to the original enol (E*) fluorescence band. The protonation of the imino group also suppresses ESIPT and sole E* emission is obtained. We disclose that chemical substitution can significantly tune the radiationless deactivation of the enol related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond rotation of the ESIPT centre. While there is a significant barrier for the experimentally studied compounds we have found a strong correlation between the barrier height and the electron donating strength of the phenyl substituent. Strong donors such as amines facilitate the barrierless non-radiative decay from E* back to the ground state, while weak electron donors make the barrier sufficiently high to allow ESIPT. Strong electron accepting groups such as –NO2 further increase this barrier. This work therefore illustrates the fine interplay necessary to design dual emitters.
N bond rotation of the ESIPT centre. While there is a significant barrier for the experimentally studied compounds we have found a strong correlation between the barrier height and the electron donating strength of the phenyl substituent. Strong donors such as amines facilitate the barrierless non-radiative decay from E* back to the ground state, while weak electron donors make the barrier sufficiently high to allow ESIPT. Strong electron accepting groups such as –NO2 further increase this barrier. This work therefore illustrates the fine interplay necessary to design dual emitters.
 Please wait while we load your content...
Please wait while we load your content...

