
Temperature dependence of dynamic, tunnelling and kinetic isotope effects in formate dehydrogenase†

Abstract
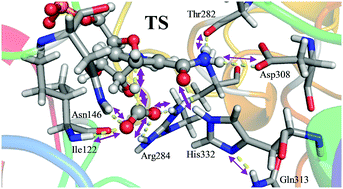
The origin of the catalytic power of enzymes has been a question of debate for a long time. In this regard, the possible contribution of protein dynamics in enzymatic catalysis has become one of the most controversial topics. In the present work, the hydride transfer step in the formate dehydrogenase (FDH EC 1.2.1.2) enzyme is studied by means of molecular dynamic (MD) simulations with quantum mechanics/molecular mechanics (QM/MM) potentials in order to explore any correlation between dynamics, tunnelling effects and the rate constant. The temperature dependence of the kinetic isotope effects (KIEs), which is one of the few tests that can be studied by experiments and simulations to shed light on this debate, has been computed and the results have been compared with previous experimental data. The classical mechanical free energy barrier and the number of recrossing trajectories seem to be temperature-independent while the quantum vibrational corrections and the tunnelling effects are slightly temperature-dependent over the interval of 5–45 °C. The computed primary KIEs are in very good agreement with previous experimental data, being almost temperature-independent within the standard deviations. The modest dependence on the temperature is due to just the quantum vibrational correction contribution. These results, together with the analysis of the evolution of the collective variables such as the electrostatic potential or the electric field created by the protein on the key atoms involved in the reaction, confirm that while the protein is well preorganised, some changes take place along the reaction that favour the hydride transfer and the product release. Coordinates defining these movements are, in fact, part of the real reaction coordinate.
 Please wait while we load your content...
Please wait while we load your content...

