
Probing the π → π* photoisomerization mechanism of trans-azobenzene by multi-state ab initio on-the-fly trajectory dynamics simulations†

Abstract
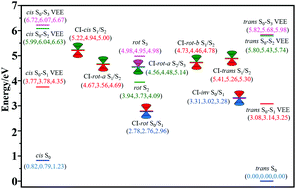
Global nonadiabatic switching on-the-fly trajectory surface hopping simulations at the 5SA-CASSCF(6,6)/6-31G quantum level have been employed to probe the photoisomerization mechanism of trans-azobenzene upon ππ* excitation within four coupled singlet low-lying electronic states (S0, S1, S2, and S3). We have performed 586 sampling trajectories (331 starting from S2 and 255 from S3), and we found about half of the sampling trajectories staying on S1 or S2 states as resonances and the other half of them ending on the ground S0 state as active trajectories. The present simulation has demonstrated that there are six distinct photoisomerization pathways which can be summarized as three categories; one is the newly opened inversion–inversion nonreactive isomerization pathway accounting for 40% (34%) of active trajectories at a time constant of 80 fs (320 fs), the other is the inversion–torsion reactive and nonreactive isomerization pathways accounting for 40% (20%) of active trajectories at a time constant of 880 fs (1700 fs), and the third is the torsion–torsion reactive and nonreactive isomerization pathways accounting for 20% (46%) of active trajectories at a time constant of 780 fs (1000 fs) upon S2 (S3) ππ* excitation. The simulated total reactive quantum yield for trans-azobenzene photoisomerization upon S2 (S3) ππ* excitation is about 0.11 (0.13) which is in good agreement with recent experimental results of 0.09–0.20. Furthermore, the newly opened inversion–inversion nonreactive isomerization pathway from the present simulation agrees well with cascade experimental measurements of the Sn → S1 → S0 relaxation mechanism in both branching ratio and time constant.
 Please wait while we load your content...
Please wait while we load your content...

