
Probing the local interface properties at a graphene–MoSe2 in-plane lateral heterostructure: an ab initio study
 a
and
a
and
Abstract
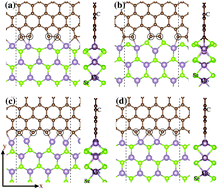
We report a theoretical study of the local interface properties at a graphene–MoSe2 (G–MoSe2) in-plane lateral heterostructure. Using a combination of first-principles density functional theory (DFT) calculations and simulations of X-ray Absorption Near-Edge Structure (XANES) spectroscopy at the C K-edge, we examined different local interface arrangements. The simulated XANES signal from interface carbon atoms showed new features compared to the pristine graphene region, which provides a way of identifying different chemical environments and/or geometries of the local interface in the G–MoSe2 lateral hybrid system. Our results also revealed that the local electronic and magnetic properties are dependent on the interface atomic structure, where metallic, semiconductor or half-metallic character was achieved at the G–MoSe2 interface. These findings indicate the great potential of 2D lateral heterojunctions for nanoelectronic and spintronic applications.
 Please wait while we load your content...
Please wait while we load your content...

