
A DFT study of RuO4 interactions with porous materials: metal–organic frameworks (MOFs) and zeolites

Abstract
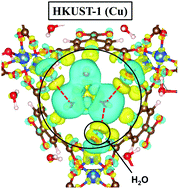
Radioactive gaseous ruthenium tetroxide (RuO4) can be released into the environment in the case of a severe nuclear accident. Using periodic dispersion corrected density functional theory calculations, we have investigated for the first time the adsorption behavior of RuO4 into prototypical porous materials, Metal–Organic Frameworks (MOFs) and zeolites, with the aim of mitigating ruthenium releases to the outside. For the MOFs, we have screened a set of six structures (MIL-53(Al), MIL-120(Al), HKUST-1(Cu), UiO-66(Zr), UiO-67(Zr) and UiO-68(Zr)), while for the zeolites two structures have been selected: mordenite (MOR) with Si/Al ratios of 11 and 5, and faujasite (FAU) with a Si/Al ratio of 2.4. The DFT calculations show that the nature of the porous materials does not have a significant effect on the adsorption energy of RuO4 compounds and that the main interaction is due to the formation of hydrogen bonds. For the tested materials, computational results show that the interaction energies of RuO4 reach their maximum with the hydrated form of HKUST-1(Cu) (−114 kJ mol−1) due to the presence of strong hydrogen bonds between the water molecules and the oxygen atoms of RuO4.
 Please wait while we load your content...
Please wait while we load your content...

